
Why Documentation Quality Defines Every Pharma Operation in Egypt
Egypt’s pharmaceutical industry was valued at USD 6.5 billion in 2024, and is expected to reach USD 13.8 billion by 2033 growing at a compound annual growth rate of 8.74%. With more than 147 export markets importing Egyptian pharmaceuticals, Egypt is home to Africa’s largest pharmaceutical market by volume and the Middle East & Africa’s second-largest pharmaceutical market by value.
Fueling that level of production starts with one often-overlooked element: data integrity in pharmaceutical documentation. Every batch produced, every test performed, every deviation justified, and every decision made needs to be documented on paper or electronically for regulators to review, audit, and rely upon.
When documentation is poor, it doesn’t just result in regulatory compliance issues. It can result in compromised product safety. For Egyptian manufacturers scrambling to meet WHO-GMP standards, Egyptian Drug Authority (EDA) regulations, and stringent regulatory requirements from export partners in Europe, the United States, and throughout MENA, faulty data management can take your business from growth opportunity to serious regulatory, operational, and commercial risk.
Here’s a look at what data integrity in a pharma documentation plant means and why it’s a critical issue for manufacturers in Egypt right now.

What Data Integrity Actually Means in Pharmaceutical Documentation
The ALCOA Framework: The Global Standard
Underlying data integrity in the pharmaceutical manufacturing industry is the ALCOA/ALCOA+ framework, which is reflected across major regulatory expectations, including FDA 21 CFR Parts 11, 210 and 211, UK MHRA’s GxP Data Integrity Guidance, WHO TRS 1033 Annex 4, and EU GMP Annex 11. Egypt’s Drug Authority also aligns its regulatory direction with WHO-GMP expectations and has applied for PIC/S pre-accession.
ALCOA Principles include:
- Attributable – who performed a task, date/time stamp and the system they worked in must be identified.
- Legible – records can be read for their entire retention period.
- Contemporaneous – data should be recorded at the time of activity
- Original – first recorded entry is the official record. Copies of the records should be able to be traced back to the original.
- Accurate – records should be factual, nothing should be omitted, added or changed
Later interpretations of regulations have expanded ALCOA to include Complete, Consistent, Enduring, and Available(ALCOA+).WHO TRS 1033 Annex 4 guideline on Data Integrity, ALCOA+ expectations apply to paper records, electronic records, and hybrid systems involved in routine pharmaceutical manufacturing operations.
Why does this apply to Egypt? Major data integrity violations observed during inspections, missing audit trails, overwriting original entries, backdating records, unrestricted access to electronic files are just some examples of the FDA’s warning letters seen worldwide. Any company exporting out of Egypt to a regulated market will be held to those same standards. Any manufacturer looking to export their products should be looking at documentation standards from a readiness-to-export perspective instead of an administrative expense.
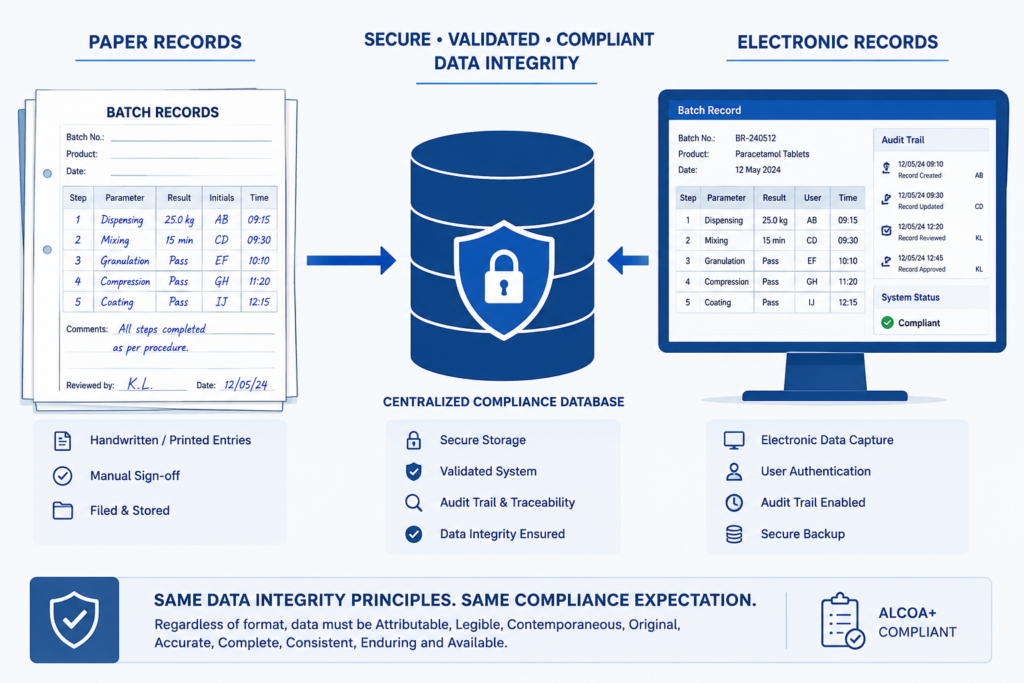
Paper Records and Electronic Systems: Different Tools, Same Standard
Egypt’s pharma industry is host to manufacturers who are at various stages of their digitisation journey. Some manufacturers are working with paper batch records; some have implemented LIMS or EBR systems. Whether records are maintained on paper or electronically, manufacturers are still held accountable for data integrity. Moving from paper to electronic systems doesn’t eliminate the requirement, it shifts how the requirement is satisfied.
- Primary controls for paper batch records include:
- Documentation is completed in permanent ink at the time of performance
- Errors are single line crossed out, initialed and dated, not erased
- Lines are used to block unused spaces
- Records are stored in a controlled, access limited environment
Technical requirements for electronic systems are defined by 21 CFR Part 11 and EU Annex 11: validated systems, audit trails enabled/reviewed, role-based access controls, use of electronic signatures where appropriate, time-stamped entries that cannot be edited without an accompanying reason-for-change.
Per WHO’s guidance, transitioning from paper-based to computerized records, or vice versa, does not remove data integrity responsibilities. Controls should follow the data.
Why Egypt’s Regulatory Environment Is Raising the Bar on Documentation
The EDA and GMP Compliance in Egypt
The Egyptian Drug Authority (EDA), Egypt’s lead pharmaceutical regulator formed in 2019, is responsible for pharmaceutical manufacturing regulations, product registration requirements, and import regulations. Egypt’s regulatory system has reached WHO Maturity Level 3 for medicines regulation, showing progress toward a stable and integrated regulatory system.
The EDA enforces GMP expectations as part of the factory licensing and registration of pharmaceutical products. Egyptian Drug Authority inspections specifically evaluate manufacturing facilities for GMP compliance. If discrepancies are found the EDA has the right to suspend or cancel the manufacturing license. The market authorisation holder is responsible for ensuring that manufacturing and distribution activities remain compliant with applicable GMP/GDP expectations as well.
Pharma Turnkey Solutions in Egypt were valued at USD 447 million in 2024. Local drug manufacture was initially primarily for the Egyptian market, but the Egyptian Government’s local-content requirements have shifted towards higher-value markets like oncology and biologics. Manufacturers wanting to export to especially Europe, the United States and regulated African countries must satisfy the documentation requirements of those regulators in addition to Egypt’s EDA. As such ALCOA+ compliance in documentation is required commercially by export dependent manufacturers.
What Inspectors Look for in Pharmaceutical Documentation
If the inspection is being conducted by EDA inspectors, WHO officials, or regulatory officials from another country performing a pre-approval or surveillance inspection at an Egyptian facility, you can expect the following documentation to be reviewed:
- Batch Records – Each step in the manufacturing process should be recorded in real time. Reviewers will be looking for entries that are completed contemporaneously, corrections that are made according to procedure, and review signatures completed prior to product release. Backdated batch records are one of the most common sources of Data Integrity observations worldwide.
- Laboratory Records – Raw data generated by instruments such as chromatographs, spectrometers, and environmental monitoring devices should be archived in its original form. If the data is transferred between computers – i.e. from the instrument to a LIMS – the process should be validated, and the audit trail should indicate that no values were altered during the transfer.
- Deviation / CAPA Records – If an issue occurs during the manufacturing process, the investigation should be documented. The root cause, corrective actions implemented, verification of effectiveness, and timeliness should all be reviewed by inspectors. Incomplete CAPA documentation is a top repeated observation in pharmaceutical inspections around the world.
- Environmental Monitoring Records – Temperature, humidity, and particulate levels from cleanroom areas should be trending, and any excursions should be investigated. Missing environmental monitoring data or excursions that were not investigated will raise questions from the inspection team.
- Training Records – Every individual who performs GMP activities should have up-to-date training records related to their job responsibilities. Incomplete training records, outdated training records, and training that does not relate to the employee’s job responsibilities are commonly cited by inspectors.
Data Integrity and Facility Design: They Are Connected
One of the more hidden correlations in pharma manufacturing is between facility design and data integrity compliance. Badly designed facilities breed data integrity hazards no quality management system can fully mitigate.
Consider how that looks from an inspector’s standpoint. A manufacturing facility where employees can traffic through cleanroom areas without properly designed airlocks, door interlocks, pressure cascades, and HEPA-filtered HVAC systems may increase the risk of excursions leading to deviations. Deviations lead to investigations. Repeat investigations because the root cause was a facility design issue and not an operational one means your CAPA is clogged with repeated observations and your quality management system is deemed ineffective by inspectors.
An entirely different documentation landscape exists for facilities designed and built to GMP from the start. Validated HVAC systems, properly classified cleanrooms, managed material and personnel flows, and qualified utility systems put you in a position where your documentation can actually do its job: accurately record a controlled process.
Connecting facility engineering to documentation quality is critical for pharmaceutical turnkey solutions for Egypt-based manufacturers. When your plant is built correctly, your documentation will reflect a controlled process. When your plant has gaps in the design, your documentation will be filled with repeated problems and unresolved investigations.
With 25+ years of experience on over 100 pharma facility projects in 18 countries, Pharma Access specializes in linking engineering design, commissioning, qualification, and validation into one seamless delivery model. Pharmaceutical consulting companies in Egypt often see similar patterns – as facility design quality increases, so does documentation quality. Approaching design from a CQV standpoint where qualification documentation is considered during the very first phase of engineering will yield a facility where batch records, environmental monitoring, and equipment logs are backed by validated, compliant systems.
Turnkey solutions for pharmaceutical companies manufacturing in Egypt face unique challenges when it comes to integrating their facility engineering with documentation and quality systems. For these companies, that is where GMP consultants in Egypt can provide the greatest value. Pharma Access’ turnkey solution includes both the physical facility itself, cleanroom design, HVAC, MEP, utilities qualification – and the quality and documentation frameworks required to support manufacturing operations day one.

Building a Data Integrity Culture: What Manufacturers Need to Do
- Data Integrity is NOT just a documentation issue. It’s a culture issue. Highlights from the WHO guidance on data integrity: many systemic weaknesses identified during inspections are linked to production pressure overriding quality requirements, inadequate quality-unit resources, and lack of management accountability for data governance.
- Here’s a great checklist for any Egyptian manufacturer who wants to shore up their Data Integrity:
- Document EVERYTHING that touches critical data –raw material testing, in-process controls, release testing – how is the data captured, recorded, transferred, stored? Where are the hybrid (paper/electronic) handoffs?
- Validate ALL computerized systems – are audit trails turned on and being reviewed? Are access controls appropriate to the role? Are time sources locked and synchronized? Is system validation up-to-date?
- Evaluate your CAPA program – are root cause analyses being performed? Are effectiveness verifications confirmed AND recorded upon completion of corrective actions?
- Train employees on DOCUMENTATION as well as GMP topics. Personnel who understand the WHY of data integrity are more likely to practice it correctly than employees who are simply told what’s expected of them. .
- Perform mock inspections – use an inspection-style checklist aligned with EDA, WHO-GMP, and international regulatory expectations when reviewing documentation. While there are numerous benefits to performing mock inspections, the gaps you identify cost you NOTHING to fix. The same gaps found during an actual inspection can cost a facility its ability to export.
FAQs: Data Integrity and Pharmaceutical Documentation in Egypt
Q1: What is data integrity and why does it matter for Egyptian pharmaceutical manufacturers?
Data integrity means that pharmaceutical records are complete, accurate, consistent, and traceable throughout their lifecycle. For Egyptian manufacturers, it matters because the EDA, WHO-GMP, and foreign regulators all require it as a condition of manufacturing authorisation and export approval. Failures in data integrity can result in product recalls, facility licence suspension, or loss of access to regulated export markets.
Q2: What are the ALCOA+ principles and how do they apply in Egypt?
ALCOA+ stands for Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, and Available. These principles define the quality requirements for all pharmaceutical records whether paper or electronic.. The WHO TRS 1033 Annex 4 guideline on data integrity ALCOA+ as the expected standard for GMP-compliant documentation in any manufacturing environment.
Q3: How do pharmaceutical turnkey projects Egypt support data integrity compliance?
A well-designed pharmaceutical plant reduces the documentation burden created by design deficiencies. When HVAC, cleanroom classification, utilities, and equipment are validated correctly from the start, environmental monitoring records, batch records, and equipment logs reflect a controlled process. Pharma turnkey solutions Egypt that include CQV-integrated design planning produce facilities where the documentation environment supports compliance from day one.
Q4: What are the most common data integrity failures found during pharmaceutical inspections in Egypt?
Common documentation failures identified during inspections of Egyptian and regional pharmaceutical facilities include batch records completed after the event, audit trails disabled or not reviewed in electronic systems, CAPA investigations closed without effectiveness verification, laboratory raw data not retained in original form, and training records that do not match the activities personnel perform.
Q5: How should an Egyptian pharmaceutical manufacturer prepare for a WHO or EDA data integrity inspection?
Map all critical data flows from generation to storage. Verify that electronic systems have validated audit trails and access controls. Review CAPA closure documentation for completeness. Conduct a mock documentation inspection. Ensure that all personnel involved in GMP activities have current, role-specific training records. Working with pharma consultants Egypt who have direct experience of WHO-GMP and EDA inspection expectations can identify gaps before they become inspection findings.



