Mumbai’s pharmaceutical sector is one of the most active in the country. And within Mumbai, Andheri West has quietly become a preferred address for pharma consultancy and engineering firms. If you are planning to set up or expand a pharmaceutical facility, finding the right pharma turnkey company in Andheri West can save you months of coordination and a great deal of money.
This post explains what pharma turnkey projects services actually cover, why Andheri West works as a base for these companies, and what you should check before signing with any firm.
What Does a Pharma Turnkey Company Actually Do?
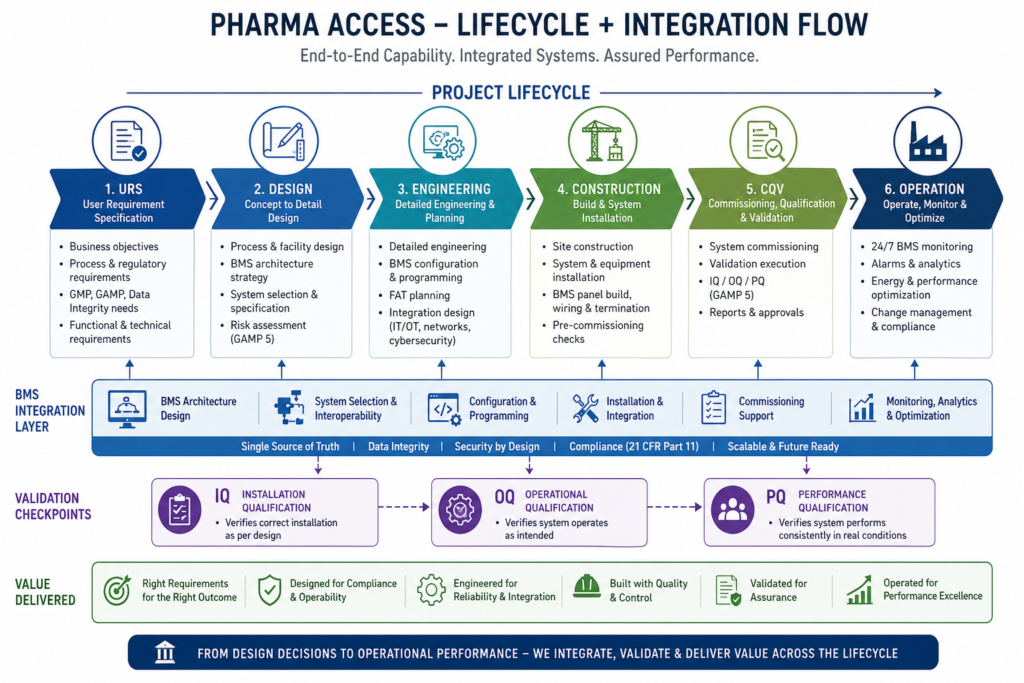
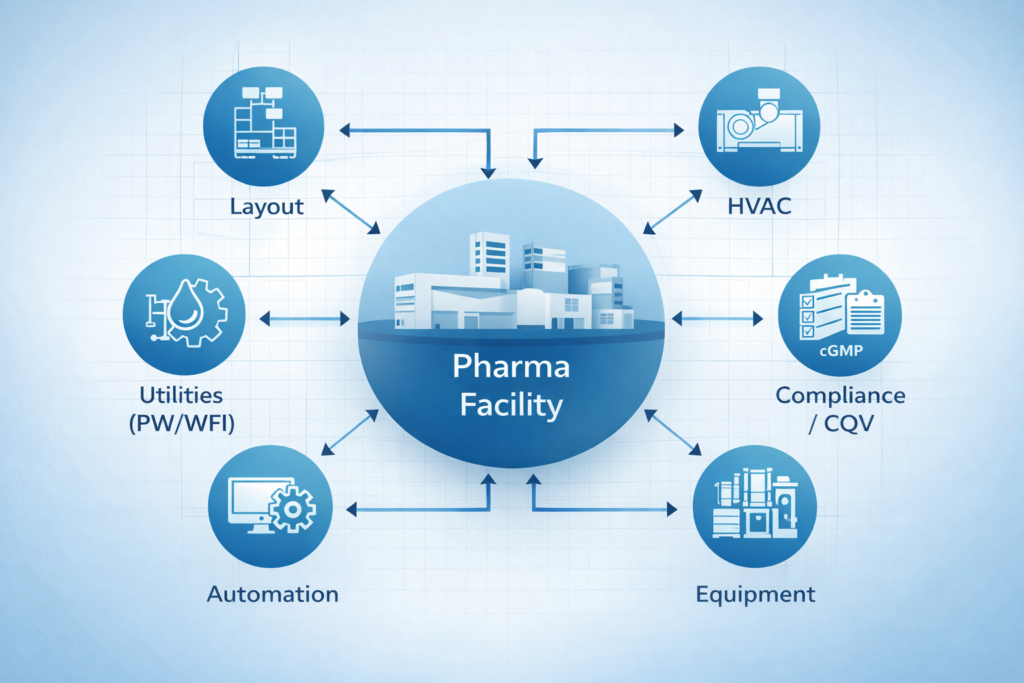
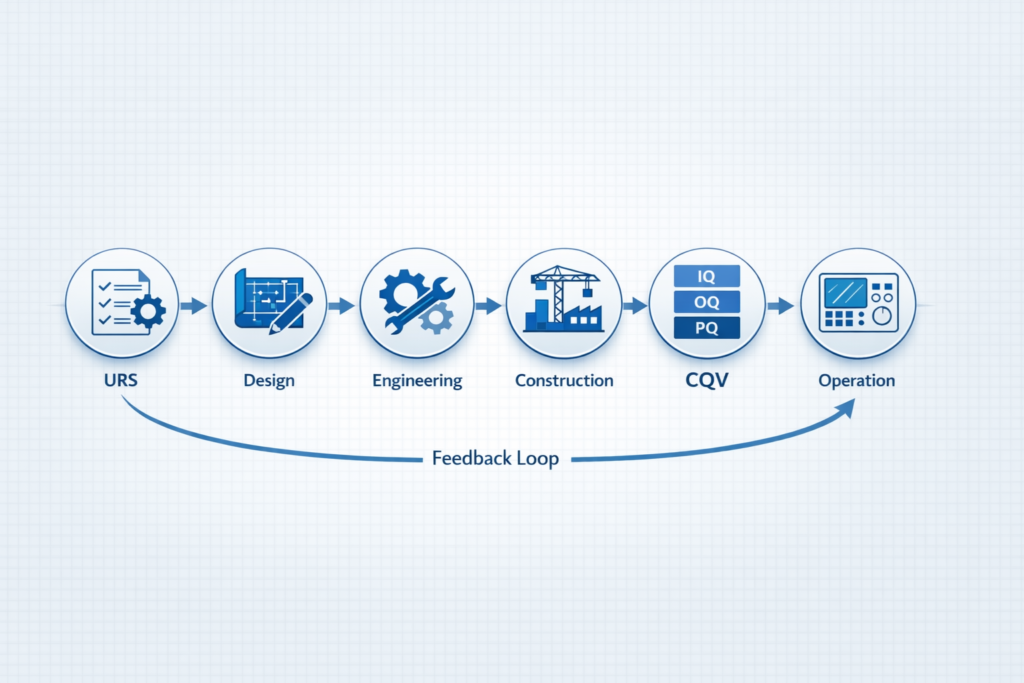
A pharma turnkey company takes responsibility for a project from start to finish. The client hands over the requirements, and the firm manages everything: engineering design, procurement, civil construction, equipment installation, and final validation.
Let’s break it down. A standard turnkey scope for a pharmaceutical facility covers:
- Engineering design: Process flow development, cleanroom classification, HVAC system design, MEP (mechanical, electrical, plumbing), piping, waste management, and automation.
- Procurement and supply: Sourcing process equipment and machinery that meet cGMP standards.
- Construction and installation: Civil, structural, and architectural work along with modular cleanroom build-outs.
- Commissioning, Qualification, and Validation (CQV): Testing and documenting that every system performs as intended before a drug product is manufactured.
- Project management: Scheduling, budgeting, vendor coordination, and regulatory documentation.
When a firm offers all of these under one roof, the client avoids coordination gaps between multiple contractors when challenges arise during project execution. The turnkey model puts accountability in one place.
Why Andheri West Works for Pharma Turnkey Firms
Andheri West is not a random choice. Here is why the location makes practical sense for a pharma turnkey company in Mumbai.
- Connectivity to pharma manufacturing clusters: Maharashtra’s major pharma production zones, including those in Navi Mumbai, Taloja, Ambernath, and Badlapur, fall within a 45 to 90-minute drive of Andheri West. Project teams can reach sites without losing half a day to commute.
- Access to talent: Mumbai has a large pool of pharmaceutical engineers, cGMP compliance professionals, and project managers. Andheri West’s position near the Western Express Highway and its metro connectivity makes it easier for firms to attract and retain qualified staff.
- Airport proximity: Chhatrapati Shivaji Maharaj International Airport sits adjacent to Andheri. For firms running projects in the MENA region, Southeast Asia, or other international markets, this matters every week. Engineers and project leads can fly out and return without losing two days to travel logistics.
- Vendor and supplier ecosystem: Andheri and its neighbouring areas in western Mumbai host a large number of industrial equipment suppliers and specialised contractors. A turnkey firm operating from here can hold supplier meetings, inspect samples, and finalise contracts without extensive travel.
- Regulatory access: Key government and regulatory offices, including those linked to the Central Drugs Standard Control Organisation (CDSCO), are reachable from Mumbai. Being in the city puts a firm closer to the regulatory conversation.
What Services Should a Good Pharma Turnkey Company Offer?
Not every firm that calls itself a pharma turnkey company delivers the same scope. Here is what a genuinely full-service firm should cover.
Engineering Design for Diverse Dosage Forms

The engineering work should go beyond generic cleanroom drawings. A capable firm should design facilities for:
- Oral solid dosage (OSD): tablets, capsules, sachets, effervescent and extended-release formats
- Sterile manufacturing: injectables including LVP, SVP, vials, ampoules, pre-filled syringes, and lyophilised products
- Oral liquid dosage: syrups, suspensions, emulsions
- Biotechnology: monoclonal antibodies, vaccines, cell and gene therapy, peptides, insulin
- Active pharmaceutical ingredients (API): covering biological routes, chemical routes, oncology, and cephalosporins
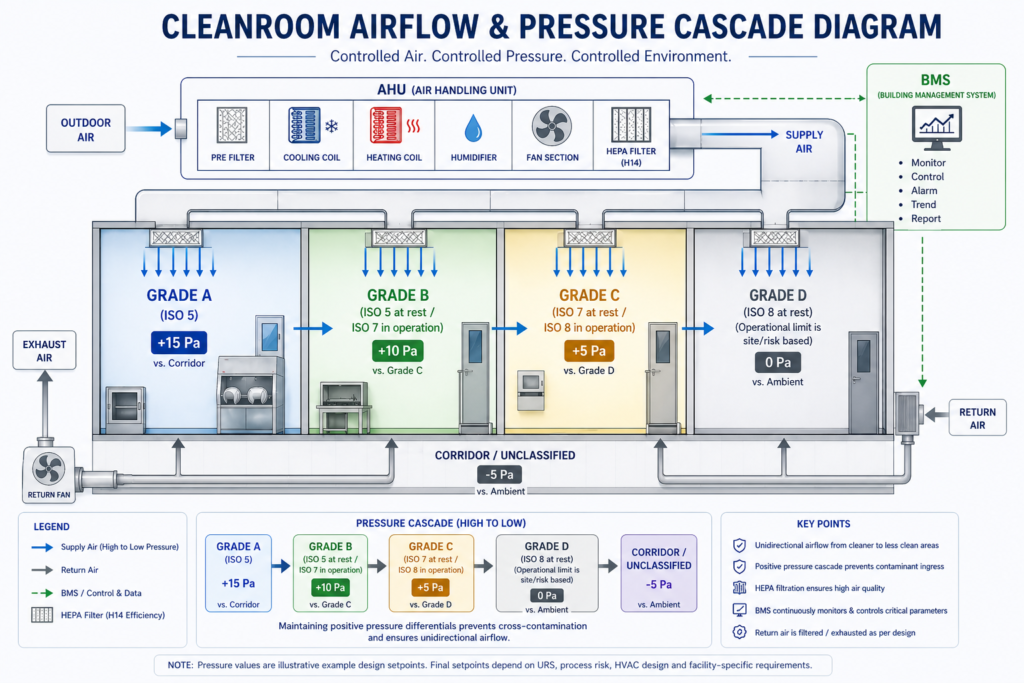
Each dosage form has a different contamination risk profile, and the facility design, HVAC classification, and pressure differentials must match those risks. A firm that has actually built different types of facilities brings a different calibre of thinking to the design table.
Regulatory Compliance Built into the Design
GMP compliance is not something you bolt on at the end. It needs to be baked into every design decision: room finishes, air change rates, material flow, personnel flow, drain placement. A firm that builds facilities for GMP-regulated environments should be familiar with WHO GMP guidelines, US FDA cGMP requirements (21 CFR Parts 210 and 211), EU GMP Guidelines, including Annex 1 where applicable, and the Revised Schedule M requirements under the Drugs and Cosmetics Rules.
The Drugs and Cosmetics Act, 1940, along with the Drugs and Cosmetics Rules and subsequent amendments, governs pharmaceutical manufacturing in India. The Revised Schedule M prescribes the Good Manufacturing Practices (GMP) requirements that pharmaceutical manufacturers are expected to follow. Any pharma turnkey firm working in India should have a thorough understanding of these regulatory requirements.
Commissioning, Qualification, and Validation (CQV)

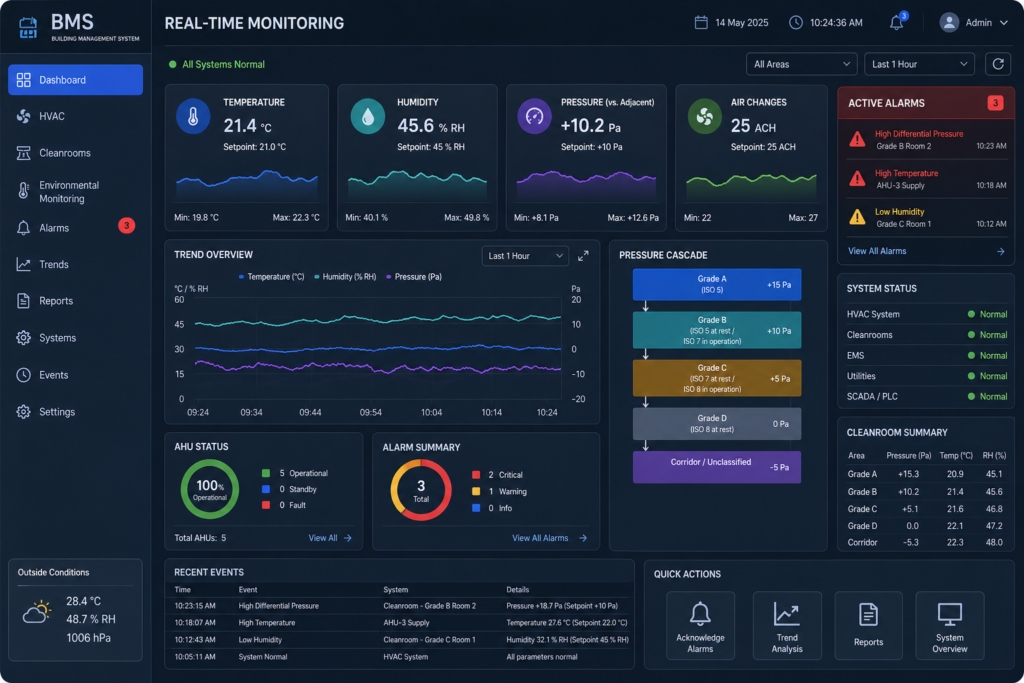
CQV is where a lot of turnkey projects go soft. Design and construction can look fine on paper, but if the firm cannot qualify the HVAC system, validate the water system, or complete the equipment qualification documentation to a regulatory standard, the client ends up hiring another consultant to fix it. A proper pharma turnkey company in Andheri West should have in-house CQV capability, not just engineering teams.
Modular and Prefabricated Facility Options
Conventional construction takes time. Modular construction, where sections of the facility are prefabricated off-site and assembled on location, cuts project timelines and reduces on-site risk. Some firms have developed proprietary modular facility concepts that allow faster installation with consistent quality.
Project Management Across Geographies
A pharma turnkey firm with a strong track record will have completed projects outside India. This matters because it signals that the firm can manage international vendor relationships, deal with customs and logistics, and coordinate multilingual teams on site.
Critical Utility Systems

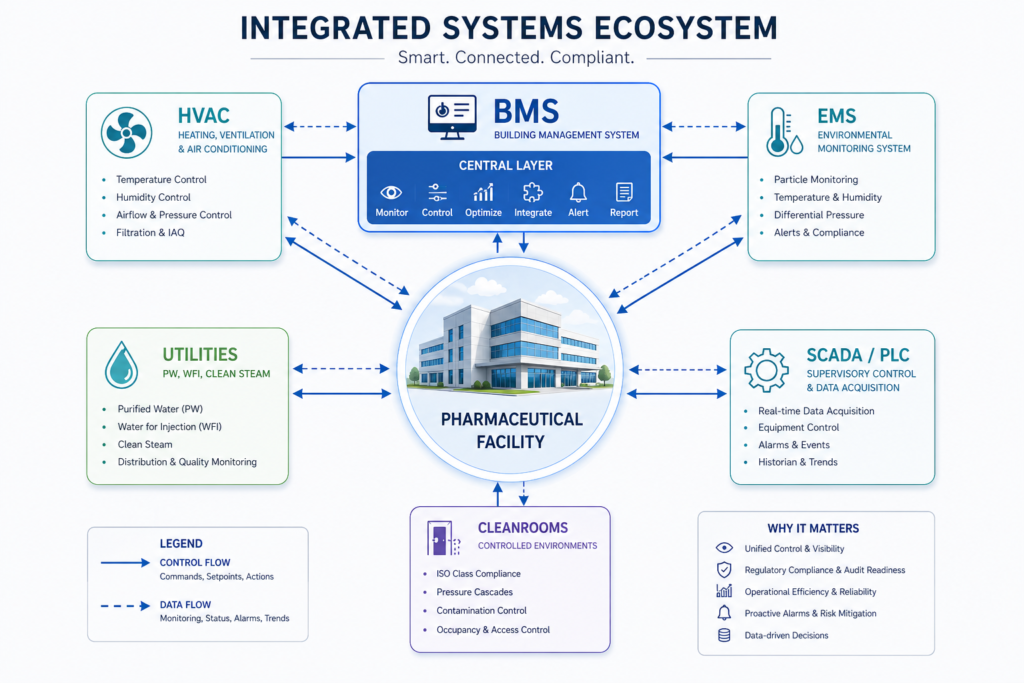
In addition to process equipment and facility construction, a pharmaceutical turnkey project should include the engineering and integration of critical clean utility systems, including Purified Water (PW), Water for Injection (WFI), Clean Steam, Compressed Air, HVAC, Building Management Systems (BMS), and Electrical & Utility Distribution Networks. These systems are essential for maintaining GMP compliance, supporting reliable manufacturing operations, and ensuring consistent product quality throughout the facility lifecycle.
Pharma Access: A Pharma Turnkey Company in Andheri West
Pharma Access is headquartered at 702, Supreme Chambers, Veera Desai Road, Andheri West, Mumbai 400053. The firm has been in pharma turnkey consulting since 2001 and has delivered over 120 projects across more than 18 countries.
Their scope spans engineering design, procurement and supply, civil construction, CQV, and project management. The firm covers all major dosage forms including biotechnology, sterile manufacturing, OSD, oral liquid dosage, and API facilities.
Pharma Access holds ISO 9001:2015 (Quality Management Systems), ISO 14001:2015 (Environmental Management Systems), and ISO 45001:2018 (Occupational Health and Safety) certifications. internationally recognised management system certifications that reflect the company’s commitment to quality, environmental responsibility, and workplace safety.
Pharma Access combines multidisciplinary engineering expertise with structured project management capabilities to support pharmaceutical projects across diverse dosage forms and regulatory environments. Their team includes over 70 engineering personnel, 12 discipline subject matter specialists, and 8 technical project managers. They also developed a Modular Mobile Facility (MMF) concept, which applies prefabricated construction to pharmaceutical environments for faster project delivery.
Clients include Dr. Reddy’s, Piramal, Aragen, Al Shifa, and projects in Algeria, Saudi Arabia, Egypt, Syria, and the Middle East.
This level of scale and geographic spread is worth mentioning because it tells you the firm has managed regulatory differences, logistics complexity, and technical challenges across different markets.
How to Evaluate a Pharma Turnkey Company Before You Hire One
Next steps matter once you start comparing firms. Here is a practical checklist.
1. Ask for project references in your specific dosage category: A firm that has done 50 OSD plants but never handled a sterile injectable facility is not the right partner for your injectables project. Ask specifically.
2. Check the CQV capability: Request their standard CQV plan and ask who on the team holds the relevant qualifications. If they plan to subcontract the validation work, find out who to and what their quality standards look like.
3. Review their regulatory track record: Have they built facilities that successfully passed WHO inspections, USFDA audits, or EU GMP assessments? Ask for documented outcomes, not just claims.
4. Understand their project management method: A firm running multiple projects simultaneously needs a structured project management system. Ask how they track schedule, budget, and change orders across active projects.
5. Visit a completed facility if possible: Nothing replaces a site visit. Ask the firm to arrange a walk-through of a recently completed project. A completed facility often provides better insight into engineering quality, workmanship, and project execution than any proposal or presentation.
6. Confirm their ISO certifications are current: ISO 9001, 14001, and 45001 certifications are independently audited. Ask for the current certificate and verify the issuing body.
Andheri West vs Other Mumbai Locations for Pharma Consultancy
Some pharma turnkey firms operate from Lower Parel, BKC, or Navi Mumbai. Each has trade-offs.
BKC and Lower Parel are closer to corporate headquarters and banking, but they sit further from industrial corridors. Navi Mumbai is close to manufacturing zones like Taloja and Mahape but can be harder for international travel.
Andheri West balances proximity to the airport, access to western and central industrial corridors, and availability of technical talent from nearby residential areas like Goregaon, Malad, and Borivali. For a firm running international pharma projects, this combination works better than a purely corporate address.
Frequently Asked Questions
1. What is a pharma turnkey company, and what does it include?
A pharma turnkey company manages the complete delivery of a pharmaceutical facility, covering design, procurement, construction, equipment installation, and validation. The client hands over the project brief, and the firm is accountable for delivering a ready-to-operate facility.
2. Why should I choose a pharma turnkey company in Andheri West specifically?
Andheri West offers direct access to Mumbai airport, strong connectivity to Maharashtra’s pharma manufacturing clusters, a large pool of qualified engineers, and proximity to key suppliers and regulatory offices. These factors make it a practical base for firms running both domestic and international projects.
3. What regulatory standards should a pharma turnkey firm be familiar with in India?
Any firm working on Indian pharmaceutical projects should understand Revised Schedule M requirements of the Drugs and Cosmetics Act, WHO GMP guidelines, and CDSCO requirements. For export-oriented facilities, US FDA 21 CFR Parts 210 and 211 and EU GMP Guidelines, including Annex 1 where applicable, needed.
4. How long does a pharma turnkey project typically take from design to validation?
Project timelines depend on the type of facility. A greenfield oral solid dosage plant may take 18 to 30 months. A sterile or biotech facility often takes longer, ranging from 24 to 48 months, because of the stricter design requirements, cleanroom classifications, and validation workload.
5. What is the difference between an EPC contractor and a pharma turnkey consultant?
An EPC (Engineering, Procurement, and Construction) contractor covers design, sourcing, and building. A pharma turnkey consultant may also include CQV, regulatory documentation support, and GMP-specific engineering that goes beyond standard civil and MEP work. The pharma-specific layer, including cleanroom design, HVAC validation, and process equipment qualification, is what separates a general EPC firm from a dedicated pharma turnkey company.