
The Cost of Getting It Wrong Is Not Abstract
FDA inspections continue to show that pharmaceutical facilities must maintain strong CGMP systems, and warning letters remain one of the most visible signs that compliance gaps have not been addressed. A warning letter is a serious regulatory communication that can lead to further enforcement action if violations are not corrected. A consent decree is a court enforced agreement that can stop or limit production, force third party audits, and subject the firm to case-specific financial penalties for missed corrective action deadlines. Abbott Laboratories spent close to $1 billion on a single consent decree they received in 1999. Warner-Lambert paid an estimated equally steep price to comply with a consent decree they received in 1993, the original fine was only $10 million.
You don’t need to be a mathematician to know that compliance risk in pharma manufacturing in the U.S. is not a regulatory burden issue. It can make or break your company. The real question is how do you manage compliance risk before it’s too late, and why strategic pharmaceutical project management is one of the most effective ways to do that.

Why Compliance Risk Accumulates During Projects
The majority of compliance issues discovered during FDA inspections are NOT the result of intentional wrongdoing. They are the consequence of choices made during the design, build, validation, and startup of a facility without consideration of regulatory implications or made under time constraints, with inadequate cross-functional collaboration, or without early regulatory guidance.
Pharma companies start a capital project to design and construct a new manufacturing facility or renovate an existing one. Engineering works in silos. Quality and validation are involved at the end of the process. Change control is nonexistent during construction. Qualification documents are hurriedly put together just before FDA’s pre-approval inspection. Equipment and systems not designed with qualification in mind fail to qualify. The FDA inspection uncovers deficiencies that were built into the project from day one.
Effective pharma project management breaks this cycle. It synchronizes engineering, quality, regulatory, and validation efforts from day one so that compliance requirements drive design choices, not limit options once the facility is built.
What Strategic Pharma Project Management Looks Like in Practice
Phase Zero: Compliance Embedded Before Design Begins
A compliant project starts years before the first line is drawn on an engineering blueprint. We in the industry refer to this as Phase Zero, or the project initiation phase. During this phase the scope, regulatory envelope, and risk profile for everything downstream are defined.
The best pharma project management teams in the USA do these things during Phase Zero:
Define your URS with regulatory requirements integrated as design inputs, rather than afterthoughts.
Capture FDA expectations up front via the guidance ICH Q9 Quality Risk pharma Management and ICH Q10 Pharmaceutical Quality System. Leverage risk assessments to inform and prioritise decisions about aseptic zoning, HVAC design, contamination control, and more.
Establish your validation master plan (VMP) in parallel with your engineering plan (EP), not after the EP is done.
Identify and assign qualified personnel to execute on quality assurance, calibration, and validation requirements early enough that they can provide meaningful input during design reviews.
Researchers at Pharmaceutical Technology reached the same conclusion as project managers who work on these projects every day. In order to build a facility that was designed and constructed with Good Manufacturing Practices (GMP) regulatory compliance in mind – with the lowest possible risk profile – key decisions can’t be left to later phases of the project. Decisions made later can lead to expensive retrofitting.
That’s where a pharmaceutical project management company in the USA can really add value. They know where compliance risk will fall during facility design and drive project scope with that understanding from day one.
Design Reviews as Risk Control Points
When design activity starts, project management has to begin managing each design review as a risk control milestone as well as a phase-of-project milestone. Every design decision including room classification, zone pressure differentials, HVAC capacity and redundancy, routing of utilities, placement of equipment, material flow, and personnel flow has regulatory consequences. 21 CFR Parts 210, 211 and especially 21 CFR require that facilities be designed to minimize the potential for contamination and cross-contamination, to allow cleaning and maintenance and be suitable for the intended use.
Design decisions that introduce potential contamination paths, insufficient separation of product grades, or difficult to qualify utility systems can lead to FDA observations time and time again. It is considered best pharma project management practice in the USA to have QA representatives participate in design reviews during the engineering phase of new builds or renovations. Quality engineers can evaluate drawings vs cGMP requirements. Design features that would cause problems during qualification are caught and corrected during engineering, when it is inexpensive to fix, rather than during qualification, when it is costly and schedule impacting.
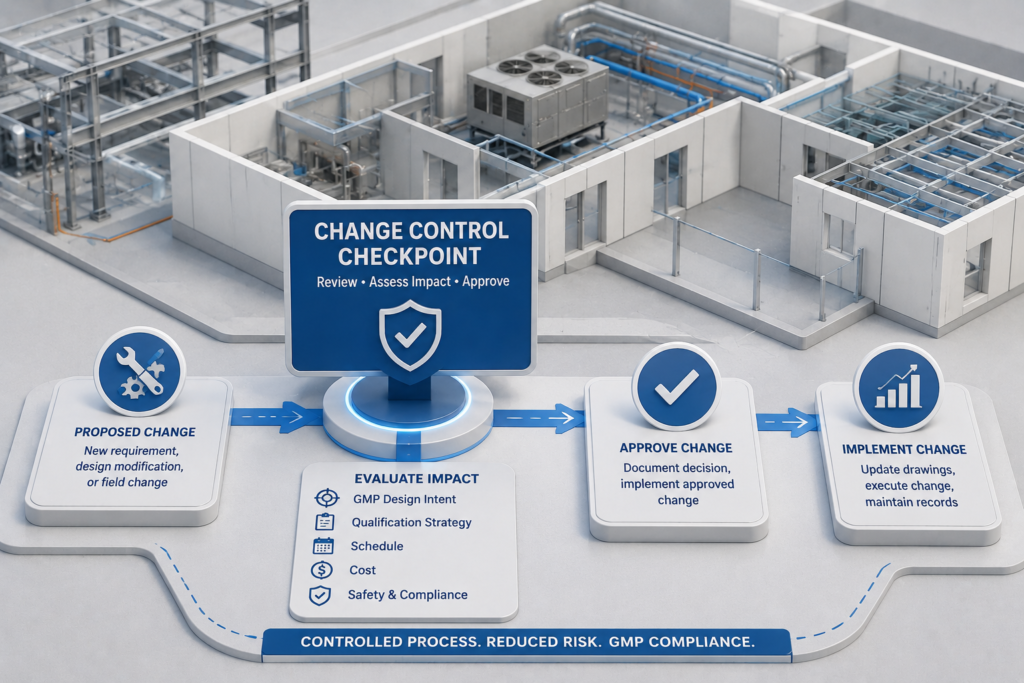
Change Control During Construction
One commonly overlooked contributor of compliance risk in pharmaceutical facility projects is failed change control during construction. Engineering changes occur on every construction project. On a pharmaceutical facility project, each and every engineering change must be analyzed for impact to the GMP design intent, qualification strategy, and future validated state of the facility prior to implementation of the change.
This requires the project management team to have an active change control process in place during construction. Whether the change is to an HVAC unit, a cleanroom surface finish, a utility connection point, equipment specifications or room layouts; the change must be reviewed through a formal, documented process including quality input and validation impact analysis. Changes requested verbally or informally without written documentation are setting the stage for non-compliance discoveries during FDA inspections.
Project teams that manage construction change control as a quality management function instead of just a cost/schedule management function will deliver a project that comes out of construction with no invisible deviations to fix prior to qualification.
CQV: Where Project Management and GMP Compliance USA Requirements Meet
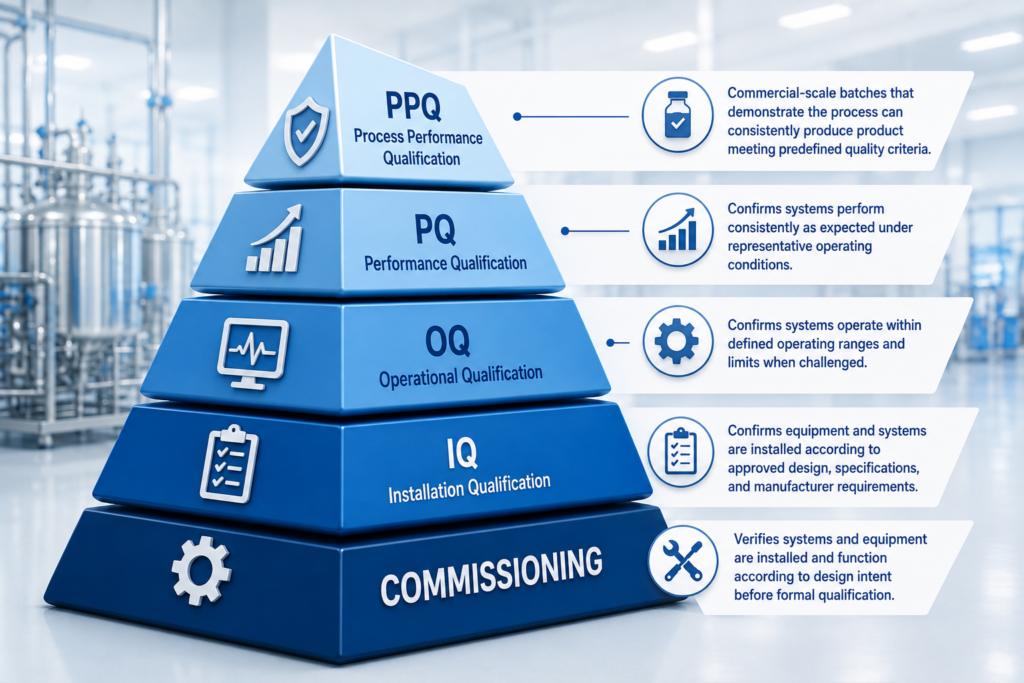
It is during Commissioning, Qualification, and Validation (CQV) when the quality of all prior project decisions is realized. Risk associated with regulatory compliance also comes to a head during this stage if previous stages have not been executed with regulatory output considered.
Here is what project teams are responsible for during CQV during a pharmaceutical facility project:
- Commissioning – ensures installed systems and equipment operate according to design intent, resulting in documented evidence of proper installation and operation.
- Installation Qualification (IQ) – ensures equipment and systems are installed as designed.
- Operational Qualification (OQ) – provides documented proof that systems operate within designed parameters when challenged.
- Performance Qualification (PQ) – provides documented proof that systems perform consistently as expected during representative operating conditions.
Process Performance Qualification (PPQ) batches made during commercial-scale manufacturing runs that prove a process can be produced consistently are an important part of FDA’s process validation lifecycle and support readiness to manufacture drug products for the US market.
Validation often becomes a project within your project. When it comes to project managing validation, Pharmaceutical Technology’s article on the topic is clear: Validation should have its own project manager, project team, project plan, and schedule that correlates with, but isn’t necessarily dictated by, the construction schedule. It also dictates that Quality Assurance and metrology teams are made aware of upcoming workloads with enough lead time to schedule resources. Lastly, the FDA district office local to your project should be made aware a new facility is being constructed.
Pharma compliance risk management teams that incorporate CQV specialists into the project delivery process from day one and treat qualification documentation as a foreseen deliverable of every design and construction decision will build facilities where regulatory filings and pre-approval inspections are predictable.
Specific Risk Zones and How Project Management Addresses Them

Data Integrity and Documentation Control
FDA has continued to increase its scrutiny on data integrity at manufacturing sites in in recent years. Data integrity gaps may include undocumented spreadsheets, missing audit trails, uncontrolled user access, incomplete electronic records, or deleted laboratory results without proper investigation. Data integrity risk starts long before IT creates a corrective action plan. It starts during project delivery when computer system validation is scoped, when decisions are made on what laboratory instruments to purchase, when specifying and implementing electronic batch record systems.
A pharmaceutical specific project management firm with USA based teams working on facility projects brings data integrity into scope during the procurement process. This ensures systems chosen and installed during construction have audit trails, validated access controls and time locked electronic records before qualification starts.
CAPA Systems and Quality System Readiness
FDA inspectors commonly cite issues such as failure to qualify suppliers properly, failure to timely root cause product failures or complaints, or inherent breakdowns in CAPA programs as systemic control failures. They are quality system issues, and they are observed when the FDA visits your pharma facility no matter how well constructed the building may be.
Project Management USA, At Strategic pharma projects, quality system readiness is one of the deliverables of the project. Developing and approving SOPs, training people on those SOPs, defining CAPA/deviation management processes, qualifying the QMS itself and preparing complaints, batch record, deviation, and change control workflows. These things all have to be planned, resourced and executed before the first batch made for regulatory submission is manufactured. Pharma Access is a turn-key pharma engineering and project management company with 25+ years of experience on over 100 projects in 18 countries.
We have made the integration of these efforts a fundamental project delivery discipline. We view the requirements for GMP compliance and the engineering execution as parallel lines driving toward the same goal – a facility ready to produce, inspect and supply.
The Business Case for Getting This Right
In plain language, the FDA’s deputy commissioner said it best: “If you think your company needs FDA inspectors to tell you where you are not in compliance, you’ve made a very expensive mistake.” This stance is supported by hard facts. Investing in pharmaceutical compliance US with effective project management discipline up front through Phase Zero planning, design reviews, construction change control and integrated CQV and quality system readiness is a fraction of the cost of remediating problems found during an inspection. Companies who receive consent decrees spend money they would have used for research and development on compliance monitoring and remediation. Some are sold off or bought out as a direct result. Schering-Plough’s first fine under a consent decree was $500 Million.
The risk of non-compliance pharma companies face is not solely technical in nature. It’s also organizational and built into how projects are planned, staffed and governed. A pharmaceutical project management company US leader that introduces regulatory strategy into the project management function from Phase Zero is not an overhead expense. It’s a risk management investment that can be measured.
FAQs
Q1: What is a pharmaceutical project management company and why does it matter for US FDA compliance?
A pharmaceutical project management company coordinates engineering design, procurement, construction, qualification, validation, and quality system delivery for pharmaceutical facility and manufacturing projects. In US FDA-regulated environments, project management that integrates compliance requirements from the beginning reduces the risk of design deficiencies, documentation failures, and qualification delays that generate FDA observations and warning letters.
Q2: What are the most common compliance risks during pharmaceutical facility projects in the USA?
The most common compliance risks during pharma facility projects include late engagement of quality and validation teams, inadequate change control during construction, systems not designed with qualification in mind, data integrity gaps in electronic systems, and insufficient CAPA and documentation infrastructure at manufacturing startup. All of these are addressable through structured pharma project management USA approaches applied from Phase Zero.
Q3: How does GMP compliance USA get built into facility design through project management?
GMP compliance is built into facility design through early involvement of quality assurance engineers in design reviews, use of ICH Q9 Quality Risk Management frameworks to assess design decisions against contamination and contamination control risks, validation master planning during the engineering phase, and construction change control that evaluates every design modification for its impact on the validated state of the facility.
Q4: What is the financial impact of pharmaceutical compliance failures in the US market?
The financial impact of pharmaceutical compliance failures includes direct costs fines of $15,000 per day under consent decrees, product recall costs, facility shutdown expenses, and third-party consultant fees and indirect costs including lost product approvals, revenue loss during remediation, reputational damage affecting partner and investor relationships, and diversion of R&D funding into compliance remediation. Historical consent decrees have cost individual companies up to $1 billion.
Q5: How does pharma compliance risk management differ in facility projects versus ongoing operations?
In facility projects, pharma compliance risk management focuses on decisions made during design, procurement, and construction that determine the regulatory baseline the facility starts from cleanroom design, HVAC qualification, utility validation, data systems integration, and quality system setup. In ongoing operations, risk management shifts to maintaining that baseline through environmental monitoring, CAPA governance, change control, periodic reviews, and inspection readiness. Both depend on quality being built in from the start rather than tested retrospectively.



