It’s no small feat to build a pharmaceutical manufacturing facility from the ground up. Before the first batch can be released for production, you will need compliant engineering designs, specialized equipment, validated utilities, and a facility that can pass the regulatory inspections. This is exactly why turnkey pharmaceutical projects have become the preferred path for drug manufacturers and biotech companies all over the world.
Let’s break it down so you know what this model actually covers, how it functions in practice, and when it makes sense for your project.
What Does “Turnkey” Mean in Pharma?

“Turnkey” is a term for construction and project delivery. A turnkey contract is when one party designs, procures, builds, and hands over a facility that is ready for GMP operation within the agreed scope. The owner just “turns the key.
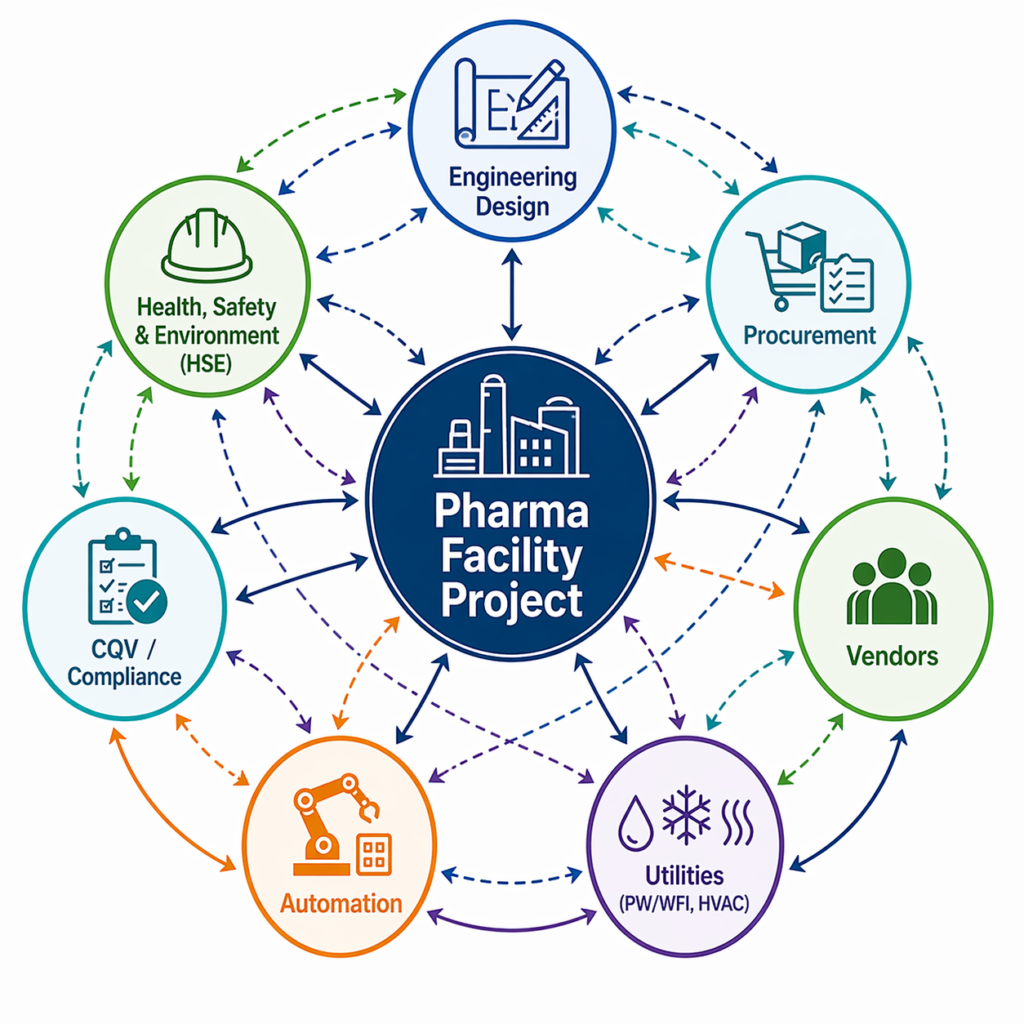
In the pharmaceutical industry, it gets several layers deeper. Turnkey pharmaceutical projects include civil and structural construction, process engineering, cleanroom design, HVAC systems, piping, electrical systems, equipment procurement and commissioning, and qualification and validation (CQV).within the agreed scope is coordinated by the contractor until the facility is inspection-ready, while the manufacturer remains responsible for GMP operations and regulatory approval.
In many cases, this model is delivered through an Engineering, Procurement, and Construction (EPC) contract. The contractor is required to provide a facility to the client at a fixed price, within a fixed time, and to a specified performance level. Otherwise, the contractor usually faces penalties in terms of the contract. This moves a large part of the project risk away from the owner.
Why this matters in pharma: Drug products marketed in the United States must be produced in facilities that meet current Good Manufacturing Practices (cGMP), required under U.S. Food and Drug Administration (FDA) regulations. The World Health Organization (WHO) also publishes its own GMP guidelines for manufacturers exporting to many global and emerging regulated markets. Any design flaw identified during a regulatory inspection can hold up or even prevent market authorization altogether.
What Do Turnkey Solutions for Biotech and Pharma Actually Include?
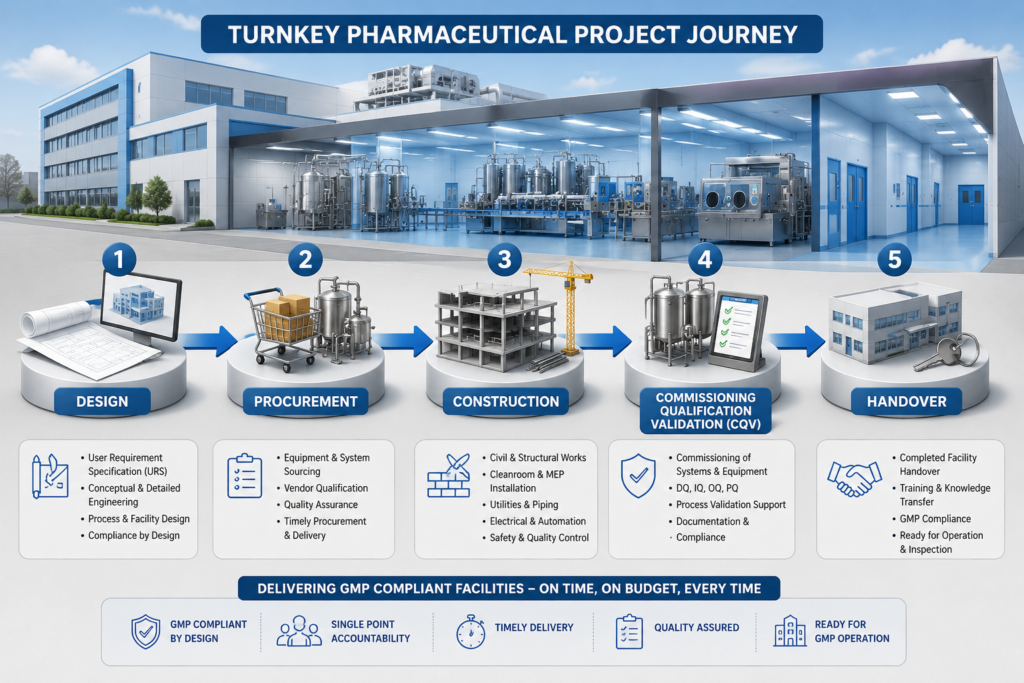
Now that we’ve established what ‘turnkey’ means in pharma, let’s examine the full range of solutions available for biotech and pharmaceutical projects. Construction is only one part of a true turnkey pharmaceutical project. Let’s go through what a full scope looks like.
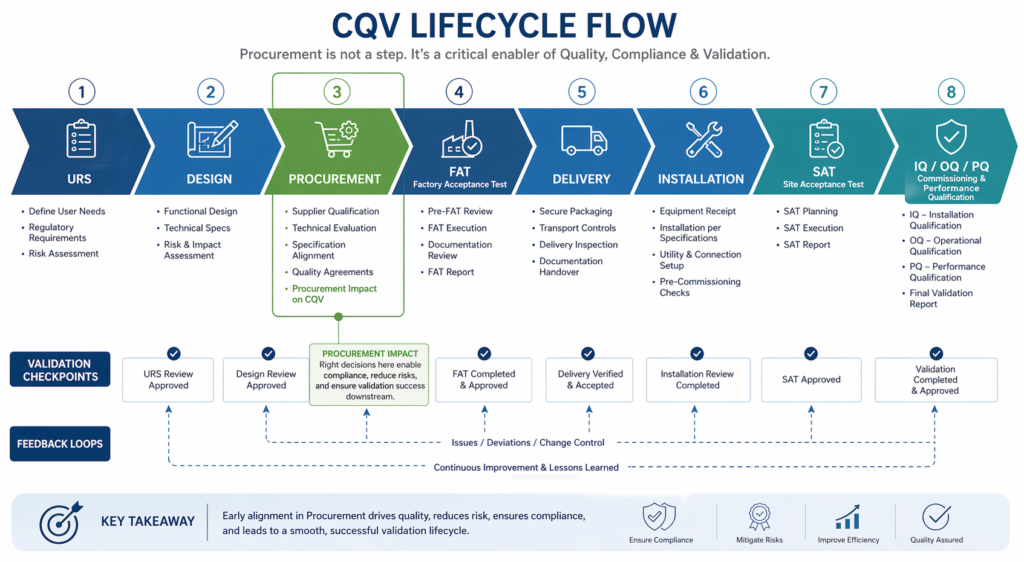
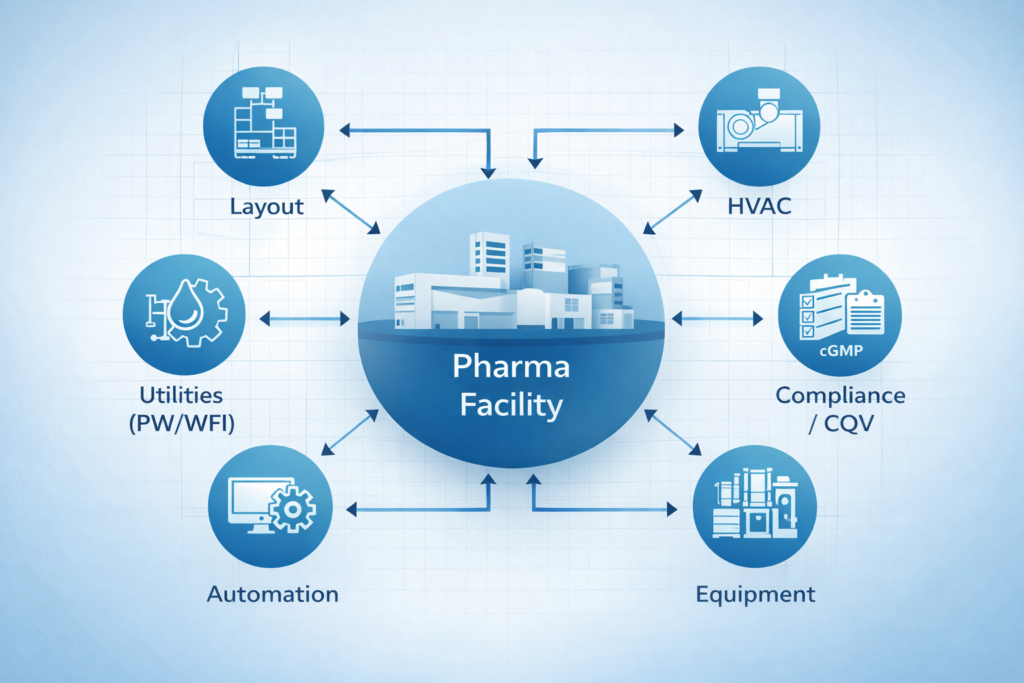
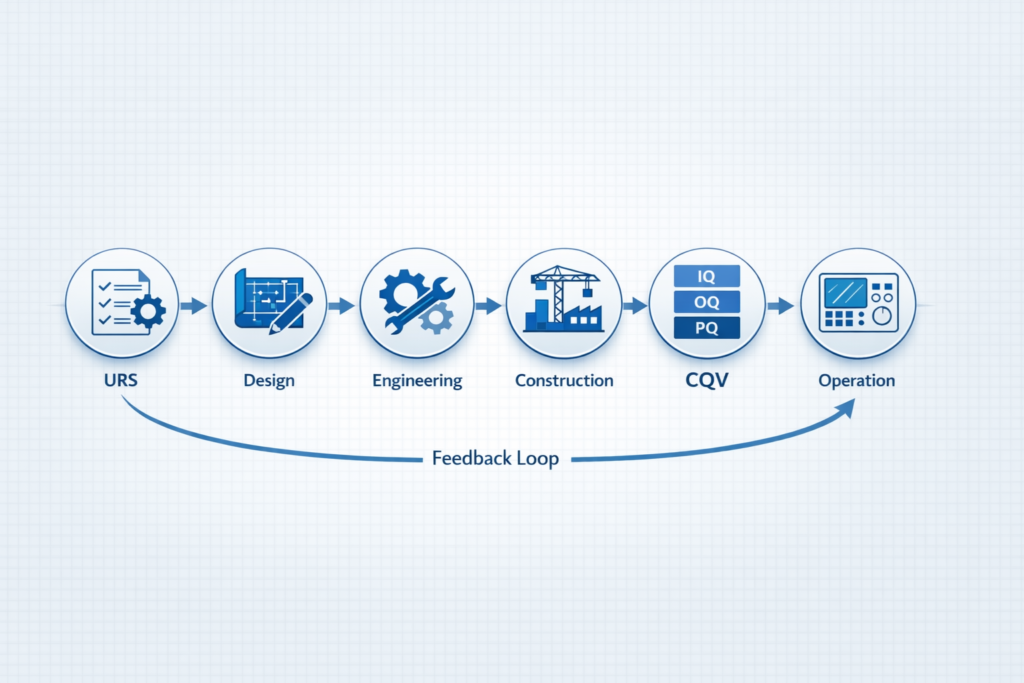
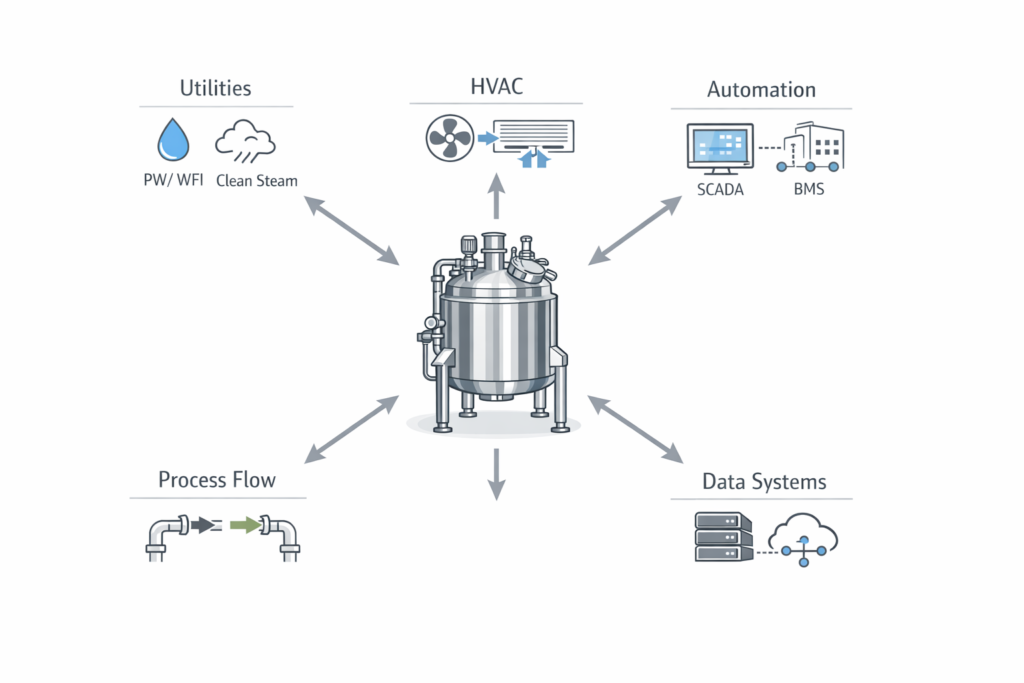
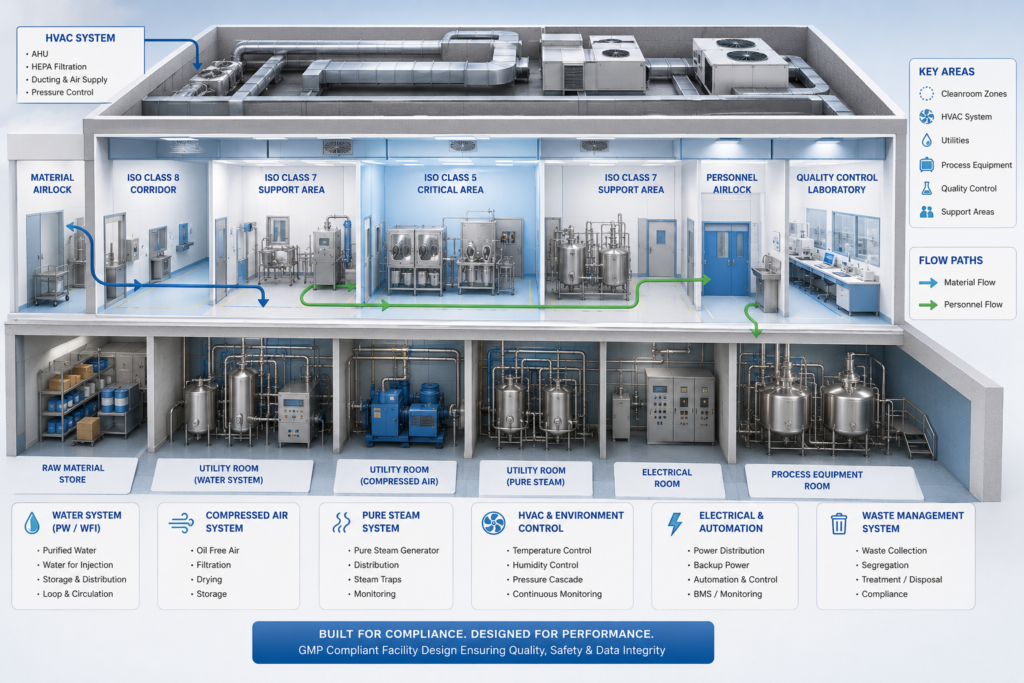
- Engineering Design: This is where it all begins. supports the preparation or refinement of a user requirement specification (URS), conceptual design, basic engineering, and detailed engineering drawings. For pharma, this means cleanroom classification layouts; HVAC design for temperature and humidity control, pressure cascades; MEP (mechanical, electrical, and plumbing) systems; and process flow diagrams.
- Procurement and Supply of Equipment: The contractor provides the sourcing and procurement of manufacturing equipment, process vessels, packaging lines, laboratory instruments, and utility systems, including purified water (PW) and water for injection (WFI). For biotech pharma facilities that often include bioreactors, fermenters, chromatography skids, and fill-finish lines.
- Construction and Installation: This phase includes civil work, cleanroom panel installation, HVAC ductwork, electrical distribution, and piping. All work shall be performed in accordance with Good Engineering Practice (GEP) principles, applicable ISPE guidance, and project-specific GMP requirements.
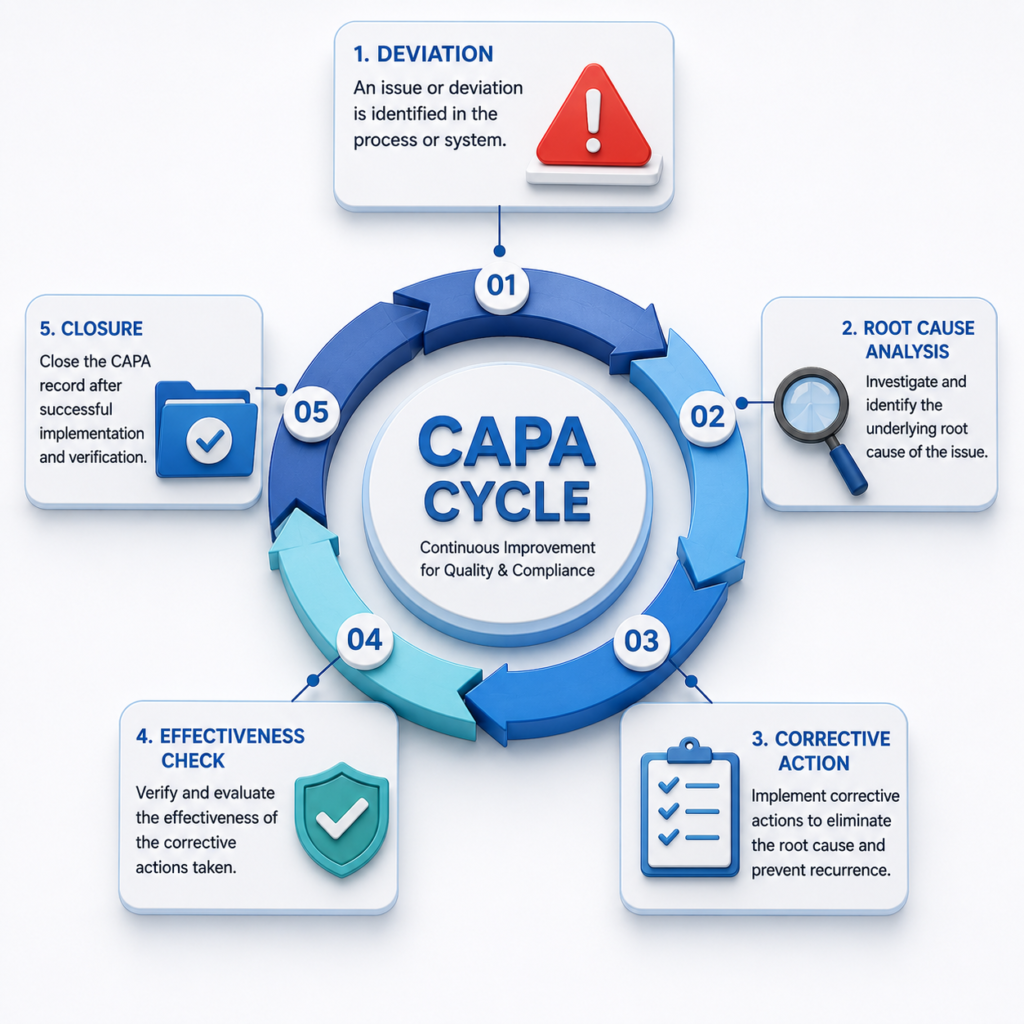
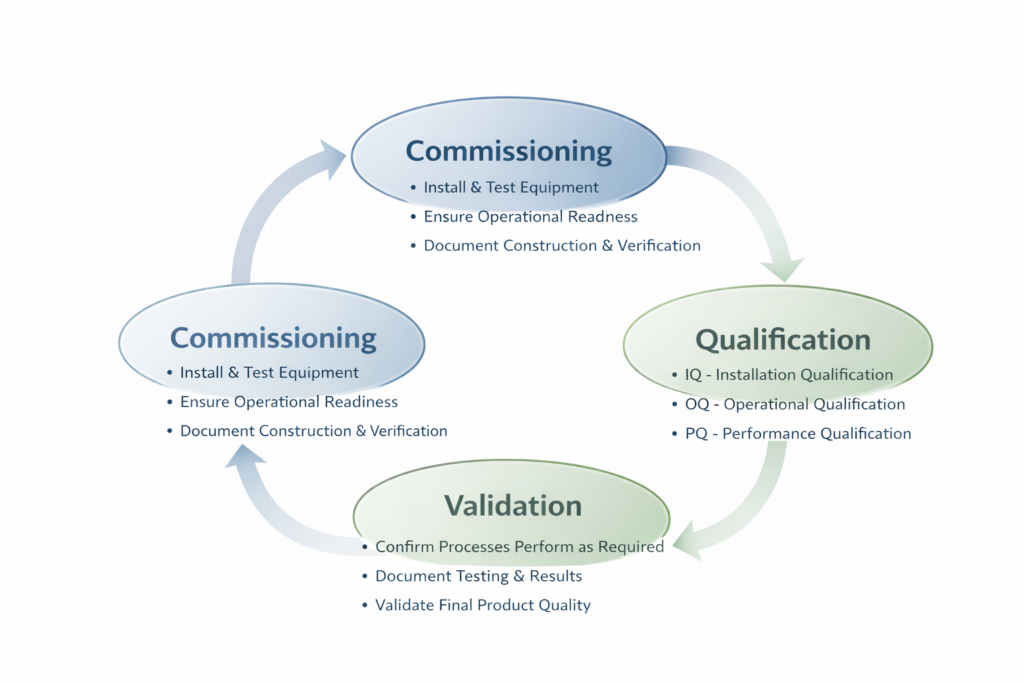
- Commissioning: Qualification, and Validation (CQV). This is the step that makes pharmaceutical turnkey projects different from normal industrial construction. Prior to any drug product manufacturing, the equipment and systems need to be Design Qualified (DQ), Installation Qualified (IQ), Operationally Qualified (OQ), and Performance Qualified (PQ), as applicable. CQV documentation is the foundation for inspection readiness, regulatory submissions, and GMP handover.
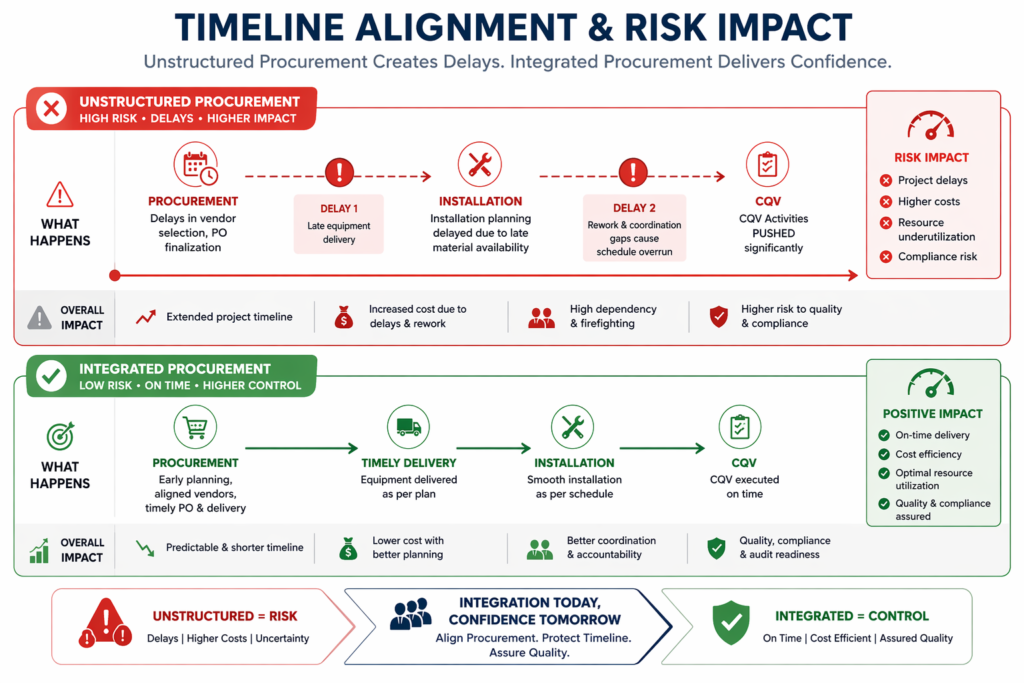
- Project Management: A turnkey delivery model has one firm overseeing all of the above. Schedule management, vendor coordination, risk tracking, and regulatory documentation are the contractor’s, not the owner’s, responsibility.
A Real-World Example of a Turnkey Pharmaceutical Project
Let’s make this concrete. Here’s a scenario.
A mid-sized pharmaceutical company in Southeast Asia is looking to build a sterile injectable facility to manufacture vials and ampoules for export to regulated markets in Europe and the United States. They don’t have construction and engineering teams in-house to manage this level of technical scope. They engage a turnkey pharmaceutical engineering consulting company.
Scope includes:
- Location selection and feasibility study support
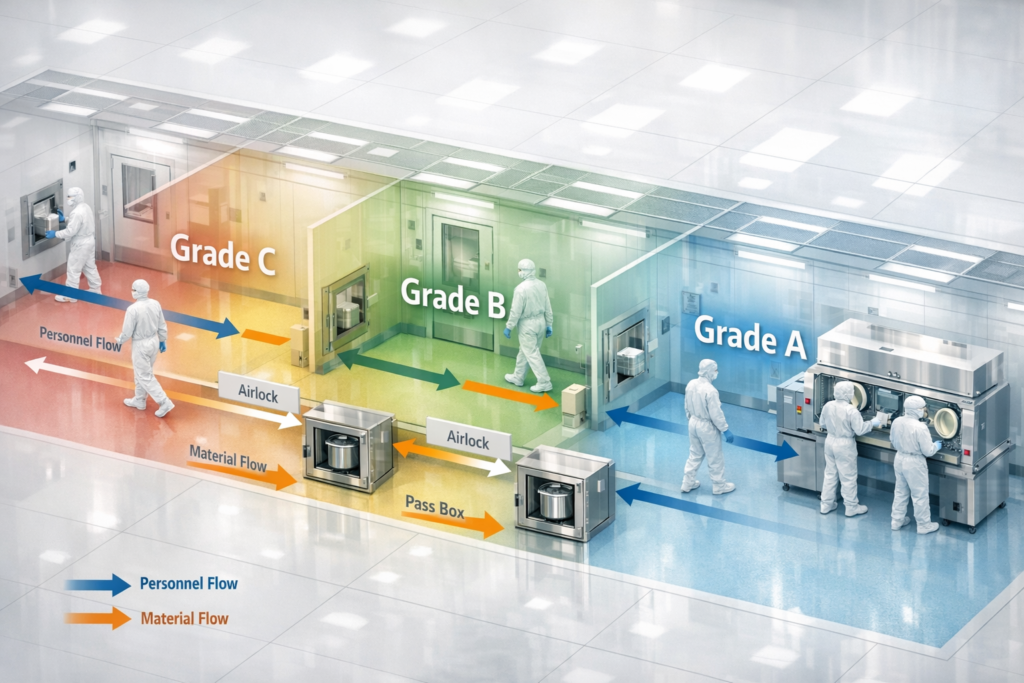
- Detailed engineering Grade A/ISO Class 5 critical filling zones, suitable ISO Class 7 support areas, and ISO Class 8 corridors, as applicable
- HVAC system design with pressure cascade control and unidirectional airflow where required in the filling zone
- Purchase of an isolator-based filling line, autoclave sterilizers, and a qualified PW/WFI system as required
- Construction of a 4,000 sq m facility block with appropriate cleanroom paneling and flooring
- Complete CQV documentation, including IQ, OQ, and PQ protocols for all process-critical systems
- Regulatory Pre-Approval Inspection Support
The project duration is about 24 to 30 months from site preparation to handover. The facility has a complete validation package at handover, and the owner’s team can start process validation and then production. The contractor has taken responsibility for design integrity, construction quality, procurement lead times, and regulatory documentation.
This is exactly what Pharma Access does. With more than 25 years of experience and over 120 projects in 18+ countries, the team at Pharma Access manages engineering, procurement, construction, and CQV under a single project umbrella for drug manufacturers building or expanding facilities across dosage forms, including sterile injectables, oral solid dosage, oral liquids, APIs, and biotech products.
When Does a Turnkey Model Make Sense?
Not all projects are worth a complete turnkey delivery. Here are the conditions in which it usually makes the best case.
- Greenfield projects with short lead times: When you are building from scratch and need a defined handover date, single-point accountability removes the coordination overhead of managing multiple engineering, construction, and validation vendors.
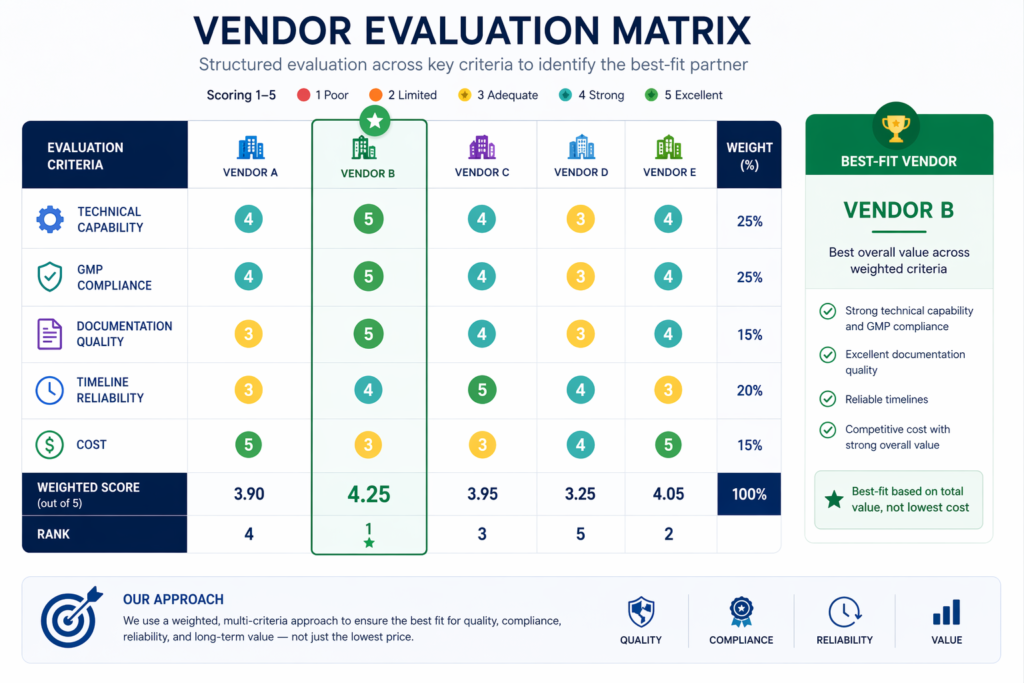
- Transnational projects: Companies that build facilities outside their home market often lack local knowledge of construction norms, procurement channels, and regulatory requirements. That gap is bridged by a pharma engineering consulting firm with regional experience.
- Biotech and advanced therapy facilities: The specialized engineering knowledge required to build cell and gene therapy (CGT) facilities, monoclonal antibody plants, and vaccine manufacturing lines is not something most general contractors have. Biotech pharmaceutical projects demand a partner with expertise in biology and engineering to deliver turnkey solutions.
- Projects under regulation: In this case, the pragmatic benefit of having a contractor who owns the build and the qualification documentation is valuable, especially if the facility needs to be ready for an FDA Pre-Approval Inspection (PAI) or a WHO audit in a defined time frame.
How Pharmaceutical Turnkey Projects Differ from Standard EPC
The standard EPC contracts for oil, gas, and power generation are centered on mechanical completion and functional testing. Pharmaceutical turnkey projects include an additional layer not found in other industries: regulatory qualification.
Deliverables such as validation of equipment and utilities against written protocols, support for GMP documentation, computerized system validation based on intended use, applicable 21 CFR Part 11 controls for electronic records and electronic signatures, and preparation of site master files should be achievable by a competent pharmaceutical EPC firm.
This is the difference between pharmaceutical engineering consulting firms that provide turnkey services and a general contractor. They have process engineers, validation specialists, regulatory affairs consultants, and quality systems professionals, as well as civil and structural engineers.
This approach is dubbed “Engicution” by Pharma Access, a portmanteau of engineering and execution. This reflects the idea that the design intent and on-site delivery need to stay aligned through all phases of the project, instead of being handed between different teams.
FAQs About Turnkey Pharmaceutical Projects
Q1. What is the difference between a turnkey pharmaceutical project and a design-build project?
Both models use a single firm for design and construction. The difference is in scope. A turnkey pharmaceutical project is more comprehensive, as it includes equipment procurement, utilities, and regulatory qualification documentation, resulting in a facility ready for GMP operation and inspection support, not just a complete structure.
Q2. How long does a typical pharmaceutical turnkey project take from start to handover?
Timelines depend on facility size and dosage form. A small oral solid dosage facility might take 18 to 24 months. A sterile injectable or biotech plant typically takes 24 to 36 months. The CQV phase itself takes six to 12 months, depending on the number of systems that need qualification.
Q3. Can a turnkey pharmaceutical project include both new construction and expansion of an existing facility?
Yes. Turnkey delivery works for greenfield (new) builds, brownfield expansions at an existing site, and upgrades within operating plants. Brownfield projects have to consider operational continuity while construction and installation are ongoing. The scope and approach are different.
Q4. What regulatory standards does a pharmaceutical turnkey project need to meet?
It depends on what markets you want to target. Facilities supplying the US market must meet FDA cGMP requirements. Many emerging-market regulators refer to WHO GMP principles or national GMP guidelines aligned with WHO expectations. EU GMP (EudraLex Volume 4) applies to European markets. Design and validation should be planned by a qualified pharmaceutical engineering consulting firm using the most rigorous applicable standard.
Q5. How do I choose the right pharmaceutical turnkey contractor for my project?
Look for a company that has hands-on experience with your dosage form and target market. Ask to see their past projects, request samples of CQV documentation, and confirm they have in-house validation and regulatory affairs capabilities. Pharma Access has a good track record in managing international regulatory environments with experience in 18+ countries with clients like Dr. Reddy’s, Piramal, and Aragen.